Getting more activated vitamin D inside the cell
Getting more activated vitamin D inside the cell
Claude AI Deep Research June 2026
The goal is not higher blood calcitriol — it is more 1,25(OH)₂D inside the target cell. In autoimmune and other extrarenal disease, the vitamin D that matters is made and used locally — inside immune cells and tissues — not the hormone the kidney releases into the blood. The kidney's output is clamped by calcium, PTH, and FGF23, so you cannot safely push it (you hit hypercalcemia first). The intracellular pool follows different rules, and those rules give you several levers.
This page treats the problem as a simple mass balance:
Intracellular 1,25(OH)₂D = (substrate delivered into the cell) × (activation rate) − (destruction rate)
Three control points follow directly: get substrate in, convert it, and stop it being destroyed.
Lever 1 — Substrate supply (the dominant lever)
This is the single most important point, and it is the one most "vitamin D booster" supplement lists leave out.
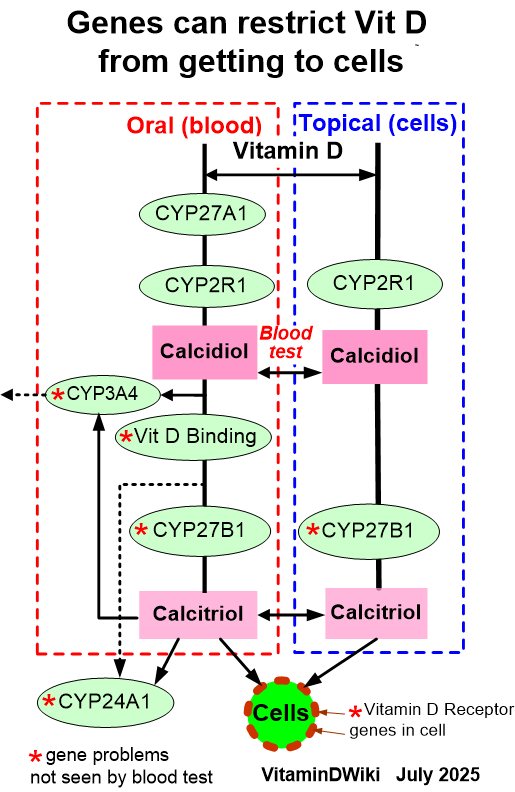
The extrarenal 1α-hydroxylase (CYP27B1) found in immune cells and many tissues behaves completely differently from the kidney's:
- It receives 25(OH)D substrate mainly by passive diffusion, which depends on a steep concentration gradient across the cell membrane.
- Its production of 1,25(OH)₂D rises as serum 25(OH)D rises and is not held back by the feedback loops that cap the renal enzyme.
In plain terms: the kidney's calcitriol output is regulated; the cell's local production is substrate-driven. Supply more 25(OH)D and the cell makes more active hormone.
The macrophage literature shows how substrate-limited this is. In TLR-activated macrophages, microbial killing is achieved more efficiently with the prohormone 25(OH)D than with finished 1,25(OH)₂D — i.e., the response tracks the host's 25(OH)D status, not circulating calcitriol.
A practical threshold exists. Two randomized trials in CKD using extended-release calcifediol suggest serum 25(OH)D needs to reach ≥ 50 ng/mL (125 nmol/L) to meaningfully engage extrarenal CYP27B1 (measured indirectly by PTH suppression). PTH-lowering response did not depend on kidney function, implying comparable delivery of 1,25(OH)₂D to intracellular VDR despite very different kidney status.
Implication: the first lever is simply getting 25(OH)D high enough — and calcifediol raises 25(OH)D faster and more predictably than cholecalciferol, bypassing the liver hydroxylation step.
Lever 1b — Free vs. bound substrate
Because uptake is diffusion-driven, it is the free (non-DBP-bound) fraction of 25(OH)D that actually crosses into the cell. Adequate free, non-DBP-bound 25(OH)D at the mitochondrial enzyme is described as the key element of the intracrine response. This is the practical content of the free-hormone hypothesis: DBP concentration and Gc genotype change how much of a given serum 25(OH)D level reaches the enzyme. Not easily druggable, but it explains why two people at the same blood level can differ.
Lever 2 — Stop it being destroyed (CYP24A1)
CYP24A1 (24-hydroxylase) is the off-switch. It degrades both 25(OH)D and 1,25(OH)₂D toward inactive metabolites (24,25D, 1,24,25D, ultimately calcitroic acid). If CYP24A1 is high, the cell shreds activated vitamin D before it can act.
Polyphenols can inhibit CYP24A1 — with a caveat
Curcumin, together with carnosic acid and silibinin, inhibits CYP24A1 expression, reducing degradation of vitamin D metabolites; the effect is Nrf2-assisted. Genistein has a similar reported effect. This is the legitimate, mechanism-specific reason curcumin belongs in this conversation — not "VDR activation."
Two caveats, flagged honestly:- The effect is cell-type-specific. Polyphenols inhibited CYP24A1 in leukemia cells but failed to — and even enhanced it — in osteoblastic cells. You cannot assume it lowers CYP24A1 everywhere.- The data are largely in vitro and in cancer models. There are no autoimmune-endpoint trials.
The FGF23 double-hit (the phosphate connection)
The systemic master-switch for these enzymes is FGF23, and it pushes both levers the wrong way at once:
- FGF23 induces CYP24A1 (more destruction)
- FGF23 suppresses CYP27B1 (less activation)
- (PTH does the opposite on both; 1,25D induces its own CYP24A1 as feedback.)
So anything that raises FGF23 — chiefly a high phosphate load, including phosphate food additives — simultaneously increases destruction and reduces activation of vitamin D inside cells. Lowering phosphate-additive intake is therefore, mechanistically, a way to protect the intracellular activated-D pool. (Cross-links: phosphate additives / FGF23 pages.)
Lever 3 — Activation-enzyme cofactors and environment
CYP27B1 is a mitochondrial cytochrome-P450 enzyme dependent on the NADPH electron-transport system and on magnesium. Magnesium is also required for all the other vitamin D-metabolizing enzymes (CYP2R1, CYP27B1, CYP24A1) and for the VDR–RXR complex to function. Magnesium deficiency throttles the entire pathway regardless of how much vitamin D is present. This is the actionable cofactor on this lever.
The local cytokine environment also gates activation: IFN-γ stimulates macrophage 1,25(OH)₂D production, while Type I interferons (IFN-α/β) and IL-4 block it. A Type-I-IFN-skewed inflammatory state — notably in lupus — can actively suppress local activation, which dovetails with the inflammation-driven VDR downregulation seen in the same diseases.
Where common supplements actually fit
Sorted by where they act on the flux, not by the loose label "VDR activator":
| Compound | Where it acts | Strength of evidence |
|---|---|---|
| Vitamin D3 / calcifediol | Lever 1 — the substrate itself (dominant) | Strong mechanistically; calcifediol pharmacokinetics well established |
| Magnesium | Lever 3 — cofactor for CYP27B1 + all D enzymes | Well established as cofactor; clinical effect on intracellular D inferred |
| Curcumin | Lever 2 — CYP24A1 inhibition (cell-type-specific) | In vitro / cancer models; bioavailability-limited |
| Reducing phosphate additives | Lever 2 — lowers FGF23 → less CYP24A1, more CYP27B1 | Mechanism solid; effect on intracellular D inferred |
| Genistein | Lever 2 — CYP24A1 inhibition | In vitro |
| Omega-3, sulforaphane, quercetin, ginger, berberine, glutathione | Indirect — lower inflammatory/IFN tone (and some lower FGF23), easing CYP24A1 induction | Convergent / indirect; little direct vitamin-D-metabolism data |
| Zinc | Downstream readout — structural for VDR's zinc-finger DNA binding | Structural requirement; not a flux lever |
Bioavailability note: curcumin, quercetin, and resveratrol are poorly absorbed in standard form; only high-bioavailability (e.g., liposomal/phytosome) preparations are plausibly relevant.
What this does NOT show
- No human autoimmune trials demonstrate that any of these levers raises intracellular activated vitamin D and thereby improves disease. The substrate logic and the ≥ 50 ng/mL threshold come mainly from CKD/PTH-suppression and TB/macrophage models; extension to autoimmune target cells is biologically reasonable but unproven.
- CYP24A1-inhibition data are in vitro and cell-type-dependent — not a guaranteed systemic effect.
- The system is self-limiting by design. 1,25(OH)₂D induces its own CYP24A1, so the intracrine pool cannot be driven away without limit. This is reassuring for safety but also caps how much "more" can be forced.
- You cannot measure the target directly. Intracellular 1,25(OH)₂D is not clinically measurable, and serum 1,25(OH)₂D does not reflect the intracrine pool. Serum 25(OH)D remains the only practical titration target.
Practical takeaway
- Raise 25(OH)D toward ≥ 50 ng/mL — the dominant lever; calcifediol if speed/reliability matters.
- Ensure magnesium sufficiency — the pathway's required cofactor.
- Protect the pool from CYP24A1 — lower phosphate-additive intake (drops FGF23); curcumin is a plausible but cell-type-specific adjunct.
- Lower the inflammatory/IFN drive — the indirect benefit of most other anti-inflammatory compounds.
Everything below substrate is a modifier; substrate plus magnesium is the foundation.
References to link
(author/journal/year as identified — verify before publishing)
- Regulation of the extrarenal CYP27B1-hydroxylase — J Steroid Biochem Mol Biol (2013/2014). Intracrine/paracrine model; IFN regulation.
- "Redefining Human Vitamin D Sufficiency: Back to the Basics" — Bone Research (2013). Macrophage intracrine schematic.
- Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase — Arch Biochem Biophys (2012). 25(OH)D more effective than 1,25D for macrophage killing.
- "Managing Dysregulated Vitamin D Metabolism in CKD" — intracrine production not feedback-limited; ≥ 50 ng/mL threshold (extended-release calcifediol RCTs).
- Plant polyphenols inhibit cellular CYP24A1 expression — AACR (Cancer Research, 2015 abstract). Curcumin / carnosic acid / silibinin; Nrf2; osteoblast exception.
- Curcumin induces cathelicidin via a VDR-independent pathway — (PubMed 22841393). Curcumin binds VDR only weakly.
- Inhibition of CYP27B1 and CYP24 increases anti-proliferative effects of 25(OH)D₃ — Anticancer Research (2021). Genistein.
- CYP24A1 / FGF23 / PTH regulation — (PMC6983441 and the CYP24A1 degradation review, Biochim Biophys Acta 2011). FGF23 ↑CYP24A1 + ↓CYP27B1.
- Uwitonze & Razzaque, "Role of Magnesium in Vitamin D Activation and Function" — J Am Osteopath Assoc (2018).
Suggested internal cross-links: calcifediol; free vitamin D hypothesis; phosphate additives / FGF23; curcumin; magnesium; CYP24A1; extrarenal activation.
Related in Vitamin D Life